Leukemia Scientific Literacy Paper
Scientific Literary Paper
Martin Ensinger
December 4, 2018
Prof: Dr. Christina Steel
BIOL 293 Section: Online
Acute myeloid leukemia (AML) is an aggressive hematologic malignancy which is caused by an accumulation of immature white blood cells (leukemia cells) in the blood and bone marrow (Minzel et al., 2018). To be diagnosed with AML, there must be at least a 20% leukemia cell count in the marrow or blood (Cancer.org, 2018). This increased leukemia cell count means the body has more white blood cells then it requires. The leukemia white blood cells are immature and not able to fight infection in the same way as mature white blood cells do. The increased number of immature white blood cells affect the function of the body’s major organs, by not having enough red blood cells in the blood to supply oxygen to the body’s systems. In addition, the blood does not have enough platelets to properly clot and not enough normal white blood cells to fight infections properly.
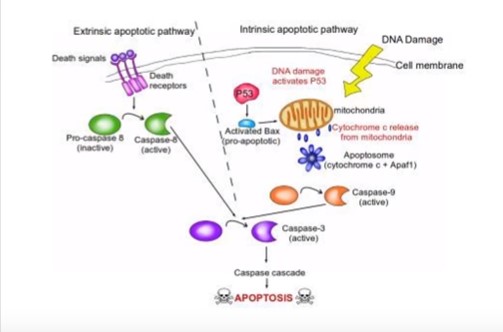
There are ten hallmarks to all cancers, one of the ten hallmarks is apoptosis or cell death. Cancer divides and grows uncontrollably by evading apoptosis. A normal cell cycle is Interphase (G1 Normal cell growth a metabolic activities, S DNA replication, G2 Growth and preparation for division) and Mitosis, Cell division. If during a cell’s normal life there some unrepairable biochemical event or change in the cell’s normal characteristic the cell initiate a programmed cell death called apoptosis (Ward, 2010).
Apoptosis can be initiated either by intrinsic or extrinsic factors. In the intrinsic pathway apoptosis is most commonly initiated by DNA Damage by breaking the double strand of DNA causing histone H2A to become phosphorylate becoming γH2AX. The γH2AX causes the activation of the p53 protein. The p53 protein is known as the guardian of the genome because it is responsible for detecting DNA damage, chromosome abnormalities, and noticing this and initiating the cell cycle to initiate repair (Levine, 1997). If repair and correction is not possible to the DNA, apoptosis is initiated by p53 activating the pro-apoptic protein BAX which initiated the release of cytochrome c from the mitochondria (McCurrach et al., 1997). Cytochrome and the release of Apaf-1 form a complex known as the apoptosome. Apoptosome causes the conversion of inactive pro-caspase into active caspase-9 and active caspase-3 which leads to the caspase cascade and apoptosis (Porter and Jänicke, 1999). Extrinsic factors, Cell Membrane death receptors signal inactive pro-caspase 5 to active capase-5. Capase-5 then activates Capase-3 and causes the caspase cascade and apoptosis. (See figure below of apoptosis sequence of events.)
The study by Minzel’s team “Small molecules Co-targeting CKIα and the Transcriptional Kinase CDK7/9 control AML in Preclinical Models.” Show that by blocking CKIα and CDK7/CDK9 will stabilize p53. Causing p53 to initiate the apoptosis death sequence in leukemia cancer cells (Minzel et al., 2018). Which was evident by H2AX phosphorylation to γH2AX, this was done without damage to the DNA. This is a new approach to the treatment in leukemia by activating p53 and the suppression of MDM2 (Levine and Oren, 2009). One of the inhibitors used in the study A51 also disrupted super enhancers. Super enhancers are a region of DNA that has multiple transcription factors that increase the likelihood of transcription and thereby blocking the transcription elongation of multiple cancer cells (Whyte et al., 2013). (see diagram’s below of interaction of proteins.)
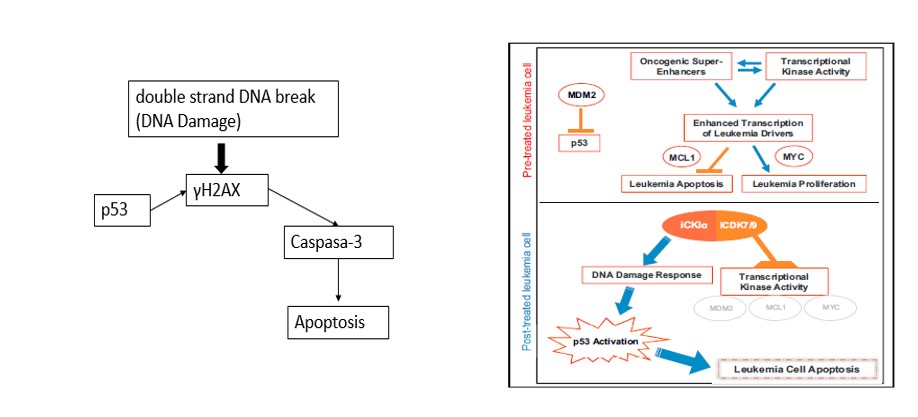
The study showed that by inhibiting CKIα causing decreasing levels β-catenin by phosphorylation. The stabilization β-catenin triggers apoptosis (Minzel et al., 2018). β-catenin is a dual function protein that is involved in the regulation and coordination of cell-cell adhesion and gene transcription and p53 by abolishing the expression of the Wingless/integrated signaling pathway (wnt). Wnt are a group of signal transduction pathways which begin with proteins that pass signals into a cell through cell surface receptors (Nusse and Varmus, 1992). The blocking of wnt is accomplished by inhibiting the catalytic sub-unit CDK9 which is the transcription elongation factor P-TEFb. And CDK7 the catalytic subunit of the transcription factor TFIIH.
In the study six CKI inhibitors were found (see below) A86 being showing the best affinity to CKIα . Though both A51 and A86 also prevent the target transcription of 2 cancer genes and one anti-apoptotic cancer gene. All inhibitors show they were able to distinguish leukemia cells from normal blood cells. Unlike genetic deletion.
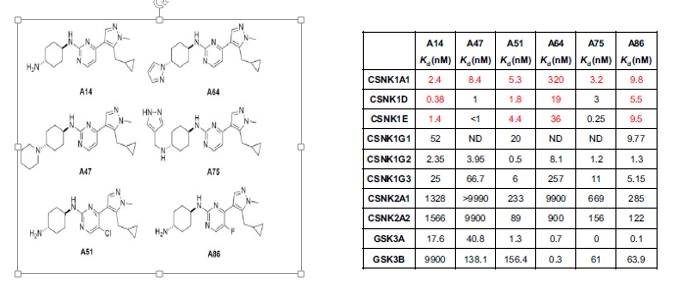
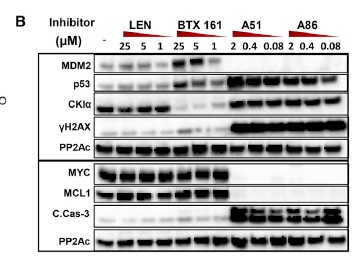
Two of the CKIα kinase inhibitors in the study also target transcriptional proteins. A51 and A86 inhibitors target 2 of the 7 known transcriptional proteins CDK7 (TFIH) and CDK9 (P-TEFb) which are considered as the gate keepers of the transcriptional machinery (Minzel et al., 2018). This is done by A51 and A86 abolishing the expression of MYC, MDM2 cancer genes and the anti-apoptotic cancer gene MCL1. mRNA analysis proved this by showing a reduction in mRNA expression of MYC and MDM2 (see Western Blot table below). Which make sense since the TFIIH and TEFb are unable to form preventing RNA polymerase II from transcribing mRNA for those genes. Also note the presence of proteins γH2AX, CKIα, p53 and Caspase-3 with inhibitors A51 and A86 in the Western Blot Table. Note that inhibitor A51 is less concentration dependent than A86.
Blocking the transcriptional protein CDK7 and CDK9. Leads to the blocking of the transcription within the following cancer genes: MYC and MDM2 and the anti-apoptotic cancer gene MCL1. Along with activating p53 and the carboxyl-terminal domain of the RNA polymerase II thus blocking transcription elongation. And activating p53 DNA Damage Response causing cell apoptosis.
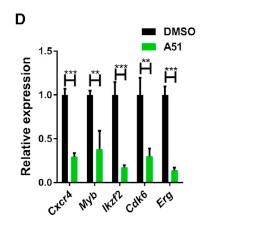
As mentioned earlier A51 also shuts down the super enhancer genes known leukemia drivers MYB, Erg, and Cxcr4. This was confirmed by mRNA analysis. (see figure below) This is thought to be done by the curtailing of P-TEFb inhibition. Which was proven by suppression downstream of the transcriptional site for the super enhancing genes and confirming the blocking of transcription elongation. (Minzel et al., 2018). This was an added benefit of the A51 inhibitor as the goal of the study was to help stabilize the p53 protein causing the cell to start the apoptosis cascade.
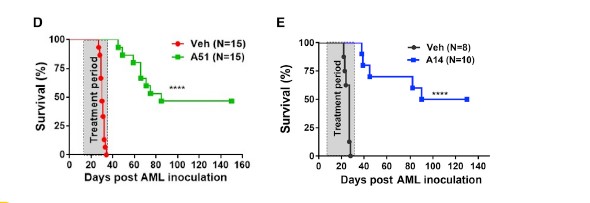
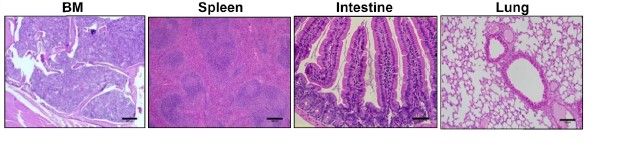
The results of the study by Minzel’s team were that forty to fifty percent of the treated mice with A51 survived and showed no signs of the leukemia up to five months post observation. Minzel’s team went a step further to prove the elimination of leukemia-propagating cells by transplanting the bone morrow from the surviving mice treated with A51 to lethally irradiated wild type mice. With no leukemia being observed after a four month period of observation (see chart and histology from surviving mice below).
Minzel’s team was successful in finding an inhibitor A51 which was capable of stabilizing p53 and causing the apoptosis cascade in cancer cells. Inhibitor A51 was less concentration dependent then inhibitor A86. Inhibitor A51 also inhibited two cancer genes Myc and Mdm2. The results of their research showed that they were able to eradicate cancer from about half the mice treated with A51 completely. Chemotherapy is 60 to 70% effective with no remission (America Cancer Society, 2018). Stem cell treatment for leukemia percentages are about 85% to 95% (Roswell Park Cancer institute, 2018). Given the cost, availability and issues people have with stem cell treatment, and the side effects of chemotherapy in treating cancer with slightly better results than the drug A51. I would recommend drug A51 in clinical trials or least recommend more research and testing on it in the treatment of leukemia.